VALIDATION4u
Definition Validation4u.
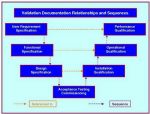
In validation4u, there cannot be a qualified “state” unless a qualified “state” has been defined. In cGMP the URS defines this qualified “state” and the execution of the DQ, IQ, OQ and PQ has to be sufficiently rigorous to verify that all the requirements detailed in the URS have been complied with.
When To Apply Qualification?
Qualification must be executed against a system or entity once it is installed, commissioned and producing product that is of the expected specification. Since all the regulatory requirements are documented, the first action must be to execute a GAP audit to establish your state of compliance. Once your GAP audit indicates you have achieved full compliance - application should be made to your regulatory authority.
Why Apply Validation4u ?
The Good Manufacturing Practice (GMP) Requirements are promulgated as regulations enforceable in law. To ensure compliance there are a powerful range of penalties that can be applied to companies and or individuals who; either negligently or willfully fail to comply with all the regulations that are applicable to their activities.
Qualification Documentation.

Full qualification is executed against a system or entity that is installed, commissioned and producing product that is of the expected specification. There must be no adjusting or tweaking to be done; that should have all been completed during commissioning activities. This is the basic methodology used to introduce pupils to the complexities of subjects like regulatory qualification.
The wording of test scripts can be tricky, but you will find that the template author has worded the essentials of the document in clear, concise and unambiguous terms allowing you to relax about such trivia and just press on following the completion instructions.
When
such documents are reviewed by regulators or visiting auditors, the quality of
the document is obvious and indicates to the reviewer the importance that the
company gives to the quality of it’s documentation.
The FDA and regulatory authorities in general, are charged with increasing the level of compliance throughout the industry. This call for increased regulatory compliance is being driven by evidence that the industry is failing to match the standards set by other regulated industries.
Our authors have designed unique interactive documents for all qualification activities such as VMP, URS, IQ.
Recent Issues.
-
How to produce superb User Requirement Specifications
Jan 25, 24 06:53 AM
 How to produce superb User Requirement Specifications is explained in the interactive lead-through SOP integral to this template.
How to produce superb User Requirement Specifications is explained in the interactive lead-through SOP integral to this template. -
Current Good Manufacturing Practice - Validation Online
Jan 20, 24 11:38 AM
 Understand Current Good Manufacturing Practice legislation and ensure your; protocols and plans are compliant with it.
Understand Current Good Manufacturing Practice legislation and ensure your; protocols and plans are compliant with it. -
How to create a winning Standard Operating Procedure
Jan 18, 24 08:36 AM
How to create a winning Standard Operating Procedure precisely replicate specific methodologies that were developed to produce a controlled product. -
How to write Retrospective Validation Protocols
Jan 18, 24 07:45 AM
How to write Retrospective Validation Protocols that will accurately certify current system compliance with URS..
Process Qualification.

Process qualification requires the establishment of scientific evidence that the process is capable of consistently delivering product that satisfies all the applicable requirements documented in the User Requirements Specification (URS).
Further to this the process validation4u should not be viewed as a one-time event, but should be viewed as an ongoing lifecycle requirement. Sampling methodology should become a key factor in carrying out ongoing process qualification.
Current good manufacturing processes (cGMP) regulations specify that samples must: represent the batch under analysis and meet specifications and statistical quality control criteria as conditions of approval and release. Furthermore, the batch must meet its predetermined specifications.
Pharmaceutical Qualification.

The importance of pharmaceutical process validation4u is emphasized by the way the regulators relate all other activities relative to it. Calibration, maintenance, documented procedures, operator training, utility and ancillary service requirements are all defined by the “process”.
Since testing for conformity can often destroy or damage the product, we are; at times, totally reliant on replicating the integrity of the process for delivery of product to the exact efficacy specified.
Process validation4u ensures that all aspects of the process execution have been subjected to rigorous verification procedures verifying that the product is consistently manufactured to specification.
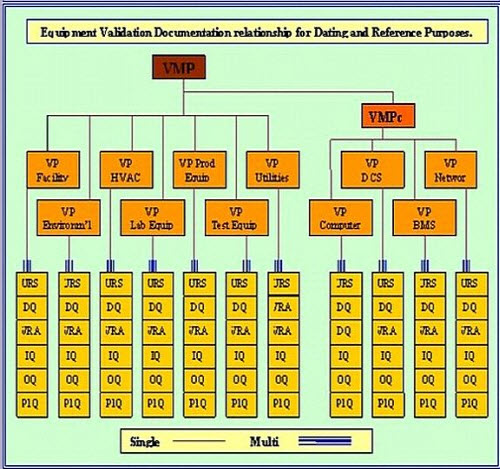
Documentation Structure.
A company approved Project Quality Plan (VP) must detail the actions, deliverables, responsibilities and scope of qualification activities required for each of the identified VRA ratings. This definition and scope must be built into each Installation Qualification and Operational qualification protocol.
Change Control.
There are two stages of Change Control to be considered, there is the equipment vendor / suppliers control used during manufacture Factory Acceptance Testing (FAT), Site Acceptance Testing (SAT) and commissioning. Then there is the post qualification control, which ensures that all work carried out on the validated system(s) is reviewed, and the impact of the work carried out on the system’s validated status is assessed.
Documented procedures.
Written procedures should be in place to describe the actions to be taken when a change is proposed to a starting material, product component, process equipment, process environment (or site), method of production or testing or any other change that may affect product quality or reproducible of the process. Change control procedures should ensure that sufficient supporting data are generated to demonstrate that the revised process will result in a product of the desired quality, consistent with the approved specifications and original validation4u.
Vendor Control.
The vendor must ensure that all changes carried out to the system during manufacture and execution of the SAT, FAT and commissioning testing, have been incorporated into the verification documentation. All drawings must be accurate, signed off, and approved. All traceability between URS documents and design specification must been maintained.
Company Control.
The company must ensure that all changes carried out to the system subsequent to the vendors actions have received approval prior to implementation, and have been fully incorporated into the system validation4u documentation. That no actions can take place that may compromise the validated state of the system.
Validation4u
21 Years of retailing cGMP - 35k templates supplied to the industry.
Combined IQ/OQ/PQ for Spreadsheets. (issue-2) $159.00
This validation online combination protocol has been specifically designed to verify that all aspects of your spreadsheet conform to best practice and that the spreadsheet layout ensures consistent and accurate use and results. The tests and inspections normally authored in separate protocols have been assembled in one protocol which is divided into three sections. This protocol enables you to verify that your developed spreadsheet application is GMP compliant, thus avoiding 483s and warning letters. You can now validate your application with minimal documentation. Equipment Validation Protocol, validation online protocol template.